Product Life Cycle Management 5i3w1m
This document was ed by and they confirmed that they have the permission to share it. If you are author or own the copyright of this book, please report to us by using this report form. Report 3b7i
Overview 3e4r5l
& View Product Life Cycle Management as PDF for free.
More details w3441
- Words: 1,436
- Pages: 27
PRODUCT LIFE CYCLE MANAGEMENT PRESENTED BY :PRINCE KUMAR M PHARMACY III SEM. 2033 SUBMITTED BY: POOJA RATHEE M.PHARMACY 2ND SEM. 1859
PRESENTED TO :DR. (MRS.) SANJU NANDA PROF. IN PHARMACEUTICS SUBMITTED TO: PROF. SANJU NANDA PHARMACEUTICS
CONTENTS: Lifecycle
management
FDA Inspection Establishment
and Enforcement
Inspection Report (EIR)
Recalls
Warning
letters
Seizures
and Injunctions 2
LIFE CYCLE MANAGEMENT:
Life cycle management is an integrated and flexible approach to business management that draws on the principles of life cycle thinking to help businesses of all kinds - manufacturers, retailers, financial, professional - understand their environmental impacts and where they occur within the lifecycle of their operation, from raw materials through to end-of-life. 3
4
FDA Inspections:
FDA developed BIMO (Bioresearch Monitoring Program) to ensure:
The protection of the rights, safety, and welfare of human research subjects
The quality and integrity of data submitted to the Agency
BIMO Program involves site visits to clinical investigators, IRBs, nonclinical (animal) laboratories, and bioequivalence analytical laboratories 5
FDA conducts inspections of clinical investigators:
Routinely to data that has been submitted to the Agency
As a result of a complaint to the Agency about the conduct of the study at the site
In response to sponsor concerns or termination of the clinical site
At the request of an FDA review division
Related to certain classes of investigational products that FDA has identified as products of special interest in its current work plan
6
Types of Inspections:
Routine inspection:
Triggered by a New Drug Application (NDA) or Pre- Market Application (PMA) submission
Routine inspections for over 80% of the inspections performed each year
Clinical Investigators who enroll the most subjects in the NDA’s pivotal trial are the most likely to be inspected.
7
For Cause Inspection: Conducting
a large volume of clinical trials
Conducting
clinical studies outside of one’s field of specialization
Reporting
significantly better efficacy, fewer adverse effects, or different laboratory results than other investigators studying the same drug
8
Inspections by Center: Center
for Biologics Evaluation and Research (CBER)
Center
for Drug Evaluation and Research (CDER)
Center
for Devices and Radiological Health (CDRH)
Center
for Food Safety and Applied Nutrition (CFSAN)
Center
for Veterinary Medicine (CVM)
9
Inspections by entity: Clinical
Investigators
IRBs Sponsors/Monitors
Good
Laboratory Practice
10
The inspector will record the following information: Dates
of IRB approvals (original, continuing review, etc.)
When
the first subject was screened
When
the first subject was consented
First
istration of the investigational product
Last
follow-up for any study subject
11
After the Inspection
The FDA inspector will hold an exit interview at the conclusion of the audit to discuss findings and deficiencies
Study staff should document the interview, specifically noting observations, comments, and commitments
Any deficiencies will be noted on the FDA form 483 and given to the PI
PI can respond to the 483 verbally during the exit interview and/or in writing 12
Post-Inspection
After the inspection, the inspector will write an Establishment Inspection Report (EIR) and submit it to FDA headquarters
After the report has been evaluated you will receive one of three letters:
No action indicated (NAI): No objectionable conditions or practices were found during the inspection, or the significance of the objectionable conditions does not justify further FDA action
13
Voluntary Action
Indicated (VAI): Objectionable conditions were found and documented, but the FDA is not prepared to take or recommend further regulatory action because the objectionable conditions are few and do not seriously impact subject safety or data integrity
Official Action
Indicated (OAI): Regulatory violations uncovered during the inspection are repeated or deliberate and/or involve submission of false information to FDA or to the sponsor 14
OAI may result in:
Warning letter (WL) Violations
can be corrected through specific action(s) by the investigator and adherence to the corrective action plan has a high probability of preventing similar or other violations in the future
Notice of Initiation of Disqualification Proceedings and Opportunity to Explain (NIDPOE) Repeatedly
or deliberately failed to comply with the requirements for conducting clinical trials
Repeatedly
or deliberately submitted false information to FDA or to the sponsor 15
Case study:
Drug firm Lupin said that it has received an Establishment Inspection Report (EIR) on April 2, 2018 from the US health regulator for its Pithampur ,Unit 1 manufacturing facility in Madhya Pradesh.
USFDA gives EIR on closure of inspection of an establishment that is subject of an FDA.
In November last, Lupin had received warning letter from USFDA for its manufacturing facilities in Goa and Pithampur.
16
RECALLS:
FDA recalls: The recall of a defective or possibly harmful product by the US Food & Drug istration (the FDA).
The guidelines of FDA categorize all recalls into one of three classes according to the level of hazard involved.
CLASS I
CLASS II
CLASS III
17
Class I recalls are for dangerous or defective products that predictably could cause serious health problems or death.
Class II recalls are for products that might cause a temporary health problem, or pose only a slight threat of a serious nature.
Example :- a food found to contain botulinal toxin, a label mix-up on a lifesaving drug, or a defective artificial heart valve.
Example:- drug that is under- strength, but that is not used to treat life-threatening situations.
Class III recalls are for products that are unlikely to cause any adverse health reaction, but that violates FDA regulations.
Example:-bottles of aspirin that contains 90 tablets instead of the 100 stated on the label. 18
The FDA develops a strategy for each individual recall that sets forth how extensively it will check on a company's performance in recalling the product in question.
For a Class I and II recall, for example, the FDA would check to make sure that each defective product has been recalled or reconditioned.
For Class III recall the FDA may decide that it only needs to spot check to make sure the product is off the market.
19
WARNING LETTERS:
An FDA warning letter is an official message from the United States Food and Drug istration (FDA) that it has found that a manufacturer or other organization has violated some rule in a regulated activity.
20
..a correspondence that notifies regulated industry about violations that FDA has documented during its inspections or investigations.
Typically, a Warning Letter notifies a responsible individual or firm that the Agency considers one or more products, practices, processes, or other activities to be in violation of the Federal Food, Drug, and Cosmetic Act (the Act), its implementing regulations and other federal statutes.
Warning Letters should only be issued for violations of regulatory significance, i.e., those that may actually lead to an enforcement action if the documented violations are not promptly and adequately corrected.
A Warning Letter is one of the Agency’s principal means of achieving prompt voluntary compliance with the Act.
21
A Warning Letter is informal and advisory. It communicates the agency's position on a matter, but it does not commit FDA to taking enforcement action. For these reasons, FDA does not consider Warning Letters to be final agency action on which it can be sued.
22
What happens when the FDA investigator arrives at the site?
The FDA investigator will:
Ask to see the top management
Present credentials (identification as an authorized FDA investigator)
Issue FDA-482 “Notice of Inspection” (explains FDA’s legal authority to inspect) 23
24
Situations where the agency will take enforcement action without necessarily issuing a Warning Letter include:
A history of repeated or continual conduct of violation. The violation is intentional or flagrant;
The violation presents a reasonable possibility of injury or death;
The violations, under Title 18 U.S.C. 1001, are intentional and willful acts that once having occurred cannot be retracted.
25
Warning letter close-out letter After the FDA completes an evaluation of corrective actions via a follow-up inspection, it may issue a so-called warning letter close-out letter if the FDA’s evaluation shows that the firm has taken corrective action to address the violations contained in the warning letter. This procedure applies to warning letters issued on or after September 1, 2009. 26
REFERENCES:
https://en.wikipedia.org/wiki/FDA_warning_letter
http://www.sustainability.vic.gov.au/Business/Efficientbusiness-operations/Lifecycle-management
https://www.fda.gov/ICECI/ComplianceManuals/Regula toryProceduresManual/ucm176733.htm#SUB6-1-1
https://www.fda.gov/ICECI/ComplianceManuals/Regula toryProceduresManual/ucm176733.htm#SUB6-1-2
27
PRESENTED TO :DR. (MRS.) SANJU NANDA PROF. IN PHARMACEUTICS SUBMITTED TO: PROF. SANJU NANDA PHARMACEUTICS
CONTENTS: Lifecycle
management
FDA Inspection Establishment
and Enforcement
Inspection Report (EIR)
Recalls
Warning
letters
Seizures
and Injunctions 2
LIFE CYCLE MANAGEMENT:
Life cycle management is an integrated and flexible approach to business management that draws on the principles of life cycle thinking to help businesses of all kinds - manufacturers, retailers, financial, professional - understand their environmental impacts and where they occur within the lifecycle of their operation, from raw materials through to end-of-life. 3
4
FDA Inspections:
FDA developed BIMO (Bioresearch Monitoring Program) to ensure:
The protection of the rights, safety, and welfare of human research subjects
The quality and integrity of data submitted to the Agency
BIMO Program involves site visits to clinical investigators, IRBs, nonclinical (animal) laboratories, and bioequivalence analytical laboratories 5
FDA conducts inspections of clinical investigators:
Routinely to data that has been submitted to the Agency
As a result of a complaint to the Agency about the conduct of the study at the site
In response to sponsor concerns or termination of the clinical site
At the request of an FDA review division
Related to certain classes of investigational products that FDA has identified as products of special interest in its current work plan
6
Types of Inspections:
Routine inspection:
Triggered by a New Drug Application (NDA) or Pre- Market Application (PMA) submission
Routine inspections for over 80% of the inspections performed each year
Clinical Investigators who enroll the most subjects in the NDA’s pivotal trial are the most likely to be inspected.
7
For Cause Inspection: Conducting
a large volume of clinical trials
Conducting
clinical studies outside of one’s field of specialization
Reporting
significantly better efficacy, fewer adverse effects, or different laboratory results than other investigators studying the same drug
8
Inspections by Center: Center
for Biologics Evaluation and Research (CBER)
Center
for Drug Evaluation and Research (CDER)
Center
for Devices and Radiological Health (CDRH)
Center
for Food Safety and Applied Nutrition (CFSAN)
Center
for Veterinary Medicine (CVM)
9
Inspections by entity: Clinical
Investigators
IRBs Sponsors/Monitors
Good
Laboratory Practice
10
The inspector will record the following information: Dates
of IRB approvals (original, continuing review, etc.)
When
the first subject was screened
When
the first subject was consented
First
istration of the investigational product
Last
follow-up for any study subject
11
After the Inspection
The FDA inspector will hold an exit interview at the conclusion of the audit to discuss findings and deficiencies
Study staff should document the interview, specifically noting observations, comments, and commitments
Any deficiencies will be noted on the FDA form 483 and given to the PI
PI can respond to the 483 verbally during the exit interview and/or in writing 12
Post-Inspection
After the inspection, the inspector will write an Establishment Inspection Report (EIR) and submit it to FDA headquarters
After the report has been evaluated you will receive one of three letters:
No action indicated (NAI): No objectionable conditions or practices were found during the inspection, or the significance of the objectionable conditions does not justify further FDA action
13
Voluntary Action
Indicated (VAI): Objectionable conditions were found and documented, but the FDA is not prepared to take or recommend further regulatory action because the objectionable conditions are few and do not seriously impact subject safety or data integrity
Official Action
Indicated (OAI): Regulatory violations uncovered during the inspection are repeated or deliberate and/or involve submission of false information to FDA or to the sponsor 14
OAI may result in:
Warning letter (WL) Violations
can be corrected through specific action(s) by the investigator and adherence to the corrective action plan has a high probability of preventing similar or other violations in the future
Notice of Initiation of Disqualification Proceedings and Opportunity to Explain (NIDPOE) Repeatedly
or deliberately failed to comply with the requirements for conducting clinical trials
Repeatedly
or deliberately submitted false information to FDA or to the sponsor 15
Case study:
Drug firm Lupin said that it has received an Establishment Inspection Report (EIR) on April 2, 2018 from the US health regulator for its Pithampur ,Unit 1 manufacturing facility in Madhya Pradesh.
USFDA gives EIR on closure of inspection of an establishment that is subject of an FDA.
In November last, Lupin had received warning letter from USFDA for its manufacturing facilities in Goa and Pithampur.
16
RECALLS:
FDA recalls: The recall of a defective or possibly harmful product by the US Food & Drug istration (the FDA).
The guidelines of FDA categorize all recalls into one of three classes according to the level of hazard involved.
CLASS I
CLASS II
CLASS III
17
Class I recalls are for dangerous or defective products that predictably could cause serious health problems or death.
Class II recalls are for products that might cause a temporary health problem, or pose only a slight threat of a serious nature.
Example :- a food found to contain botulinal toxin, a label mix-up on a lifesaving drug, or a defective artificial heart valve.
Example:- drug that is under- strength, but that is not used to treat life-threatening situations.
Class III recalls are for products that are unlikely to cause any adverse health reaction, but that violates FDA regulations.
Example:-bottles of aspirin that contains 90 tablets instead of the 100 stated on the label. 18
The FDA develops a strategy for each individual recall that sets forth how extensively it will check on a company's performance in recalling the product in question.
For a Class I and II recall, for example, the FDA would check to make sure that each defective product has been recalled or reconditioned.
For Class III recall the FDA may decide that it only needs to spot check to make sure the product is off the market.
19
WARNING LETTERS:
An FDA warning letter is an official message from the United States Food and Drug istration (FDA) that it has found that a manufacturer or other organization has violated some rule in a regulated activity.
20
..a correspondence that notifies regulated industry about violations that FDA has documented during its inspections or investigations.
Typically, a Warning Letter notifies a responsible individual or firm that the Agency considers one or more products, practices, processes, or other activities to be in violation of the Federal Food, Drug, and Cosmetic Act (the Act), its implementing regulations and other federal statutes.
Warning Letters should only be issued for violations of regulatory significance, i.e., those that may actually lead to an enforcement action if the documented violations are not promptly and adequately corrected.
A Warning Letter is one of the Agency’s principal means of achieving prompt voluntary compliance with the Act.
21
A Warning Letter is informal and advisory. It communicates the agency's position on a matter, but it does not commit FDA to taking enforcement action. For these reasons, FDA does not consider Warning Letters to be final agency action on which it can be sued.
22
What happens when the FDA investigator arrives at the site?
The FDA investigator will:
Ask to see the top management
Present credentials (identification as an authorized FDA investigator)
Issue FDA-482 “Notice of Inspection” (explains FDA’s legal authority to inspect) 23
24
Situations where the agency will take enforcement action without necessarily issuing a Warning Letter include:
A history of repeated or continual conduct of violation. The violation is intentional or flagrant;
The violation presents a reasonable possibility of injury or death;
The violations, under Title 18 U.S.C. 1001, are intentional and willful acts that once having occurred cannot be retracted.
25
Warning letter close-out letter After the FDA completes an evaluation of corrective actions via a follow-up inspection, it may issue a so-called warning letter close-out letter if the FDA’s evaluation shows that the firm has taken corrective action to address the violations contained in the warning letter. This procedure applies to warning letters issued on or after September 1, 2009. 26
REFERENCES:
https://en.wikipedia.org/wiki/FDA_warning_letter
http://www.sustainability.vic.gov.au/Business/Efficientbusiness-operations/Lifecycle-management
https://www.fda.gov/ICECI/ComplianceManuals/Regula toryProceduresManual/ucm176733.htm#SUB6-1-1
https://www.fda.gov/ICECI/ComplianceManuals/Regula toryProceduresManual/ucm176733.htm#SUB6-1-2
27
Related Documents 3m3m1z

Product Life Cycle Management 5i3w1m
December 2019 54
Product Life Cycle Management 5i3w1m
April 2021 0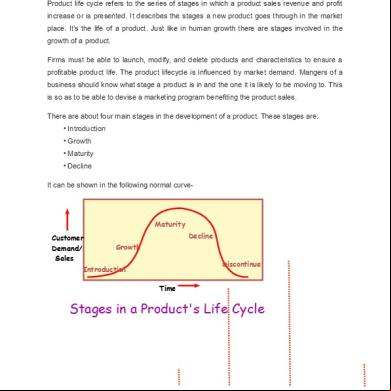
Product Life Cycle 3r4a59
November 2019 47
Starbuck Product Life Cycle j4z4c
November 2019 48
Nokia Product Life Cycle 4az6r
November 2019 120
Product Life Cycle-cadbury 1s4v6z
November 2019 67More Documents from "Deepanshu Chawla" 616nn
Product Life Cycle Management 5i3w1m
April 2021 0
Testlabz Goc Question Bank 84j3t
December 2019 138
Ftre 2014 Class Vii Answers 3o2f51
December 2019 52
Constrained Motion Problems 6z4n4t
November 2019 48
Aits 2j1d40
December 2019 76